ENGLISH|企业邮箱|OA系统|Oracle Clinical












上周,“袁来如此”专栏正式开篇,JDB电子医药子公司深圳博瑞副总经理袁智博士以《大分子生物分析概论(一):大分子药物生物分析的基础知识》为题,梳理了大分子药物分析方法的基础概念。
本期开始,“袁来如此”专栏将就LBA定量方法的监管验证展开详细介绍,预计将有多篇重磅干货内容呈现,敬请垂注!
1992年,美国弗吉尼亚州水晶城,全球首个关于生物分析方法验证会议召开,会议讨论共识的生物利用度、生物等效性和药代动力学研究的生物分析方法成为了后来制药行业进行生物分析方法验证的实际准则。史称水晶城会议。
水晶城会议还讨论了生物分析方法验证的普遍问题,同时也承认了色谱和非色谱测试方法(包括免疫测试方法和微生物方法)之间存在差异。以水晶城会议为基础,后续的行业会议和文献对生物分析方法验证进行了多次讨论。
迄今为止,人们最重视的是常规小分子药物的生物分析方法的验证,其主要原因是自从1990年以来,作为小分子药物的常规分析工具——串联式液相色谱-质谱仪(LC-MS/MS)的使用率在迅速地增长。而大分子的生物分析方法亦对此借鉴颇多。
在本文中,LBA 是免疫测试方法(immunoassays)的同义词,指任何基于大分子相互作用进行定量分析的方法;监管验证(regulatory validation)则是通过验证后方法所产生的分析数据,可供生物药物的临床前毒理研究和临床申报、注册使用。后续文章将详细解释监管验证与非监管验证之间的差异,敬请关注。
一个分析方法的生命周期通常可分为3个阶段:方法开发(method development)、研究前验证(pre-study validation)和研究中验证(in-study validation)。方法开发是建立分析方法的过程,而研究前验证是对分析方法的确认,研究中验证则是在应用过程中,如出现关键成分发生变化后,进行的部分验证。
为了确保一个分析方法能用于大分子定量,以支持药代动力学评估,并经得起监管审核,需要对特定分析方法的各个组成部分进行评估、验证和持续监控。因此,只要使用该分析方法,其验证就是一个持续的过程。
所以,方法验证是一个动态的过程。表1总结了需要评估的组分和对于分析方法生命周期中每个阶段的建议。本文的组织也对应于这些方法组分,在相应的章节中,将讨论在不同阶段中可能开展的活动的更完整、详信的信息。
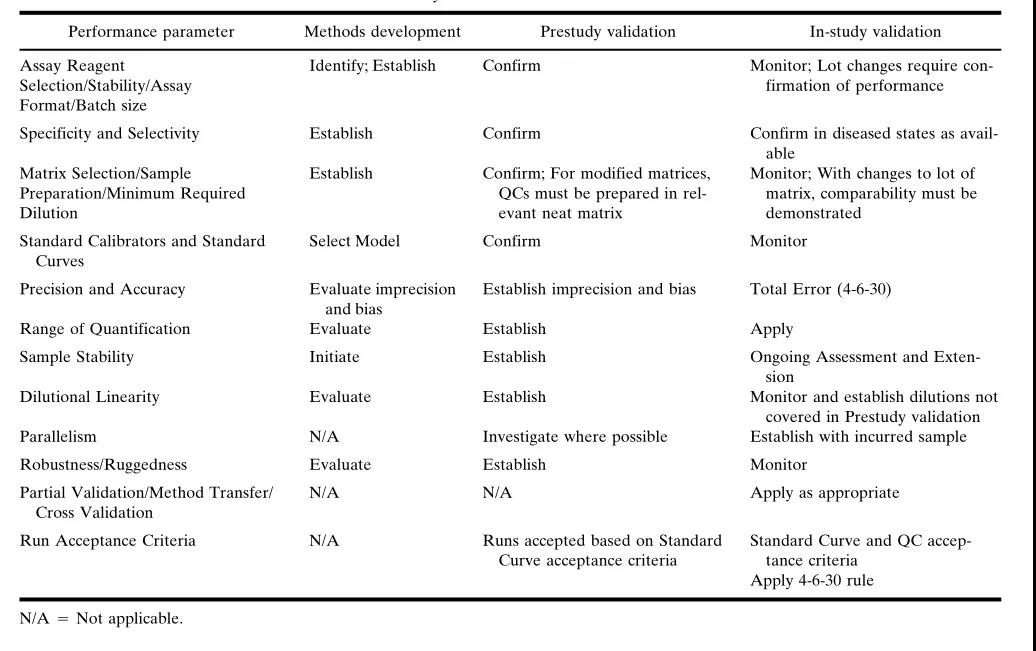
研究前验证阶段
研究中验证阶段
方法开发阶段
研究前验证阶段
研究中验证阶段
方法开发阶段
分析方法的特异性将取决于所使用的抗体或抗体对(antibody pair)的预先确定的特异性。抗体可以从商业来源获得或内部生产。无论哪种情况,在选择之前都必须评估抗体结合特性的数据,评估分析特异性的方法通常是分析样本基质,样品基质+不同浓度的待测物及其变异体、物理化学上类似的化合物以及样品基质+可能与待测物共同使用的化合物。
在某些情况下,可以使用一个样本基质来评估特异性,该样本基质包含与体内浓度相当的一种或多种与待测物有关的化合物。通常,竞争式测试格式比夹心式测试格式更易受干扰物的干扰,因为夹心式分析方法使用两种抗体,故具有更大的特异性。
如果无法获得待测物的变异体或相关形式,则可能无法在方法开发过程中生成交叉反应性(分析特异性assay specificity)数据。因此,可能需要对经过验证的分析方法的特异性进行回顾性评估,因为随着时间的流逝,将产生更多有关待测物行为的数据。
在方法开发过程中,评估选择性即是评估存在基质成分时对待测物的定量。这些基质成分可能会干扰抗体与待测物的结合,应当在定量下限(LLOQ,即低于低水平QC样品的浓度)或之上的附近外加待测物到同一样本基质类型的多个批次(至少10个)中,并评估相对误差的百分比(%RE)。尽管选择性问题通常在定量范围的低端发生,也需要在较高待测物浓度下评估选择性。在背景干扰与浓度有关的情况下,必须确定在哪个待测物的浓度之下可能出现干扰。在方法验证之前,可能需要相应地提升最低的定量限。
研究前验证阶段
研究中验证阶段
本文如有疏漏和误读相关指南和数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊、官方网络报道等公开渠道, 不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备,欢迎读者提供有价值的文献及其评估。
8. 扩展阅读
袁来如此|大分子生物分析概论(一):大分子药物生物分析的基础知识
3. C. M. Riley and T. W. Rosanke. Development of validation of analytical methods: progress in pharmaceutical and biomedical analysis (vol 3) Elsevier (Pergamon), NY 1996.
4. V. P. Shah, K et al. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetics studies. Conference Report. Eur J Drug Metabol Pharmacokinetics 16:249–255 (1991).
5. Guideline on validation of analytical procedures: definitions and terminology International Conference of Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use Geneva 1995 (1996).
6. V. P. Shah, et al. Bioanalytical method validation. A revisit with a decade of progress. Pharm. Res. 17:1551–1557 (2000).
7. K. J. Miller, et al. Workshop on Bioanalytical Methods Validation for Macromolecules: Summary Report. Pharm. Res. 18:1373–1383 (2001).
8. Guidance for the Industry. Bioanalytical Method Validation US Department of Health and Human Services FDA (CDER) and (CVM) May 2001.
9. DeSilva B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res. 2003;20:1885–900.
10. J. O. Westgard. Points of care in using statistics in method comparison studies. Clin. Chem. 44:2240–2242 (1998).
11. Watson, RG, et al. Implementing a tiered approach to bioanalytical method validation for large molecule ligand-binding assay methods in pharmacokinetic assessments. Bioanalysis. 2017 9(18):1407-1422
12. D. Rodbard, et al. Kinetics of Two-Site Immunoradiometric (Sandwich) Assays-II. Immunochem. 15:77–82 (1978).
13. B. D. Plikaytis, et al. Determination of parallelism and nonparallelism in bioassay dilution curves. J. Clin. Microbiol. 32: 2441–2447 (1994).
14. C. Hartmann, et al. Reappraisal of hypothesis testing for method validation; Detection of systematic error by comparing the means of two methods or two laboratories. Analytical Chem. 67:4491–4499 (1995).
15. S. R. Searle, et al. Variance Components Chapter 3. John Wiley & Sons, Inc, New York, NY (1992).
16. R. W. Mee. b-expectation and b-content tolerance limits for balanced one-way ANOVA random model. Technometrics 26:251–254 (1984).












